©1996-2005 All Rights Reserved. Online Journal of Pharmacokinetics. You may not store these pages in any form except for your own personal use. All other usage or distribution is illegal under international copyright treaties. Permission to use any of these pages in any other way besides the before mentioned must be gained in writing from the publisher. This article is exclusively copyrighted in its entirety to OJPK publications. This article may be copied once but may not be, reproduced or re-transmitted without the express permission of the editors.
OJPKTM
Online Journal of Pharmacokinetics ©
Volume 3: 1-15, 2005
Pharmacokinetics and hepatotoxicity of flutamide liposomes
after IV administration
Umrethia ML, Ghosh PK, Majithiya RJ, Murthy RSR
Drug Delivery Research Laboratory, Pharmacy Department, The M.S. University of Baroda, Vadodara, India
ABSTRACT
Umrethia ML, Ghosh PK, Majithiya RJ, Murthy RSR Pharmacokinetics of flutamide liposomes
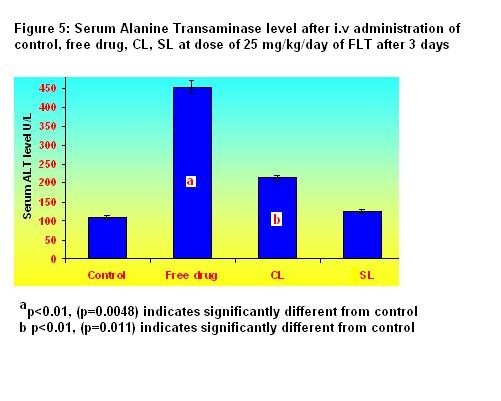
after IV and hepatotoxicity Online Journal of Pharmacokinetics, 3 : 1 -15, 2005 Flutamide (FLT) is a non-steroidal anti-androgenic used for the treatment of prostate cancer: the high doses required can be hepatotoxic. Encapsulated FLT (CL) with liposomes (SL) were thus formulatedto evaluate their blood kinetic properties, reduce RES uptake to reduce its hepatotoxicity. The size controlled FLT encapsulated CL and SL were prepared using egg phosphotidylcholine (ePC) and cholesterol by thin film hydration technique followed by high-pressure homogenization. In case of SL, methoxy polyethylene glycol 2000- phosphotidyl ethanolamine (mPEG2000-PE) was used along with lipids to provide surface hydrophiliicty to liposomes. The prepared liposomes were characterized for entrapment efficiency (EE), particle size, physical stability and percent drug retained in human plasma up to 24 hr. The CL of lower size (136 nm) with EE of 99.28% were found less stable than SL of 158 nm particle size and 98.54% EE in human plasma at 37ºC. The systemic pharmacokinetics and biodistribution of FLT encapsulated CL and SL was compared with that of free drug. The FLT-OH level in plasma and various organs of rats was determined by selective, sensitive, reproducible and validated reverse phase high-performance liquid chromatographic method. The pharmacokinetic studies showed that SL was still retained in plasma, where as CL had almost disappeared from blood circulation after 24 hr. The value of t1/2, Vss, MRT and AUC were found to be much higher for SL than that of CL and free drug. Accumulation of the FLT-OH in liver and other organ was found higher following free drug administered iv than that of CL and SL. Liver and spleen uptake of SL was 82% and 75% less than those of CL. Histopathological studies and Alanine transaminase (ALT) analysis showed that FLT encapsulated SL may help in reduction of undesired side effects associated with liver following free drug administration.
Keywords: Flutamide, liposomes, pharmacokinetics, biodistribution, histopathology
While various anti-cancer agents are used to treat patients suffering from cancers, reasons for failure of chemotherapy involves daily multi-dosing therapy for several months include drug toxicity and emergence of drug resistance (Sharma et al., 1997). Increasing their amount in tumor cells, their presence in blood and reducing their exposure to normal cells can improve their therapeutic efficiency. Thus, the current strategy for enhancing the therapeutic activities of currently available drugs is to encapsulate them inside a delivery system from where they are slowly released over extended time period (Labana et al., 2002).
Liposomal encapsulation reduces the toxicity and side effects associated with anti-tumor agents by altering their pharmacokinetics and distribution (Steerenberg et al., 1984: Herman et al., 1983: Gabizon et al., 1994: Newman et al., 1999: Gondal et al., 1993). These formulations have been used as carriers of cytotoxic drugs with the strategy based on reduction of toxicity and passive delivery to tumors (Pezer-soler, 1989: Gabizon, 1989) and improve the in vivo performance of these drugs as well as provide a repository for the drug to be released slowly so as to allow for prolonged therapeutic effect (Kondari et al., 2003) Although conventional liposomes can encapsulate a variety of drugs, they are recognized in-vivo by Kupffer cells of RES (Dijkstra, 1984) and cleared rapidly from the circulation so that a key obstacle to liposomes is to achieve control of their blood circulation and tissue distribution (Woodle, 1984). The term ‘stealth liposomes’ has been coined to designate long-circulating liposomes (Goren et al., 1997) which have a carefully selected size (about 100 nm) and are covered with covalently attached polyethylene glycol polymer (Lasic, 1995) that generates a steric barrier preventing hydrophobic interactions of plasma opsonins with the vesicle surface and inhibiting the uptake by RES (Torchilin, 1995). One key feature of long-circulating liposomes is their ability to accumulate in tumors, resulting in a significant selectivity in drug delivery to tumors and prevents its exposure to normal cells (Allen et al., 1991: Oku, 1994).
Flutamide (FLT) (3![]() -trifluoromethyl-4
-trifluoromethyl-4![]() -nitro methyl
propionylanilide) is a non-steroidal pure anti-androgen (Baker et al.,
1967) indicated for palliation of advanced prostate cancer. It acts by
inhibiting the uptake and/or binding of di-hydro testosterone to the
target cell receptor and affecting Prostate Specific Antigen (PSA)
level in blood, thus interfering with androgen action which requires
its stability in blood for enough time (Goldspiel et al., 1990). Its
pharmacokinetics and dosage characteristics (usually three doses per
day of 250 mg each) make it suitable candidate for design of controlled
release delivery systems. Flutamide is rapidly and completely
absorbed. The biologically active alpha-hydroxylated metabolite
(FLT-OH) reaches maximum plasma concentrations in about 2 hours,
indicating that it is rapidly formed from flutamide following 250 mg of
oral dose in humans Reported FLT toxicity includes diarrhoea and
hepatotoxicity
(Chabner et al., 1996).
-nitro methyl
propionylanilide) is a non-steroidal pure anti-androgen (Baker et al.,
1967) indicated for palliation of advanced prostate cancer. It acts by
inhibiting the uptake and/or binding of di-hydro testosterone to the
target cell receptor and affecting Prostate Specific Antigen (PSA)
level in blood, thus interfering with androgen action which requires
its stability in blood for enough time (Goldspiel et al., 1990). Its
pharmacokinetics and dosage characteristics (usually three doses per
day of 250 mg each) make it suitable candidate for design of controlled
release delivery systems. Flutamide is rapidly and completely
absorbed. The biologically active alpha-hydroxylated metabolite
(FLT-OH) reaches maximum plasma concentrations in about 2 hours,
indicating that it is rapidly formed from flutamide following 250 mg of
oral dose in humans Reported FLT toxicity includes diarrhoea and
hepatotoxicity
(Chabner et al., 1996).
Attempts have been made to formulate FLT liposomes and compared the pharmacokinetic, bio-distribution, histopathology and biochemical analysis (ALT level measurement) of its liposomal preparations with that of pure drug in rats specially to reduce hepatotoxicity associated with FLT as well as to increase therapeutic efficacy.
MATERIALS AND METHODS
Materials: Flutamide was kindly supplied as a gift sample by Coral Drugs Pvt. Ltd., New Delhi; India. Egg Phosphotidylcholine (ePC), Methoxy Polyethylene Glycol (M.Wt 2000) (mPEG 2000), Phosphotidylethanolamine (PE) and Cholesterol (CHOL) were purchased from Sigma Chemical Co., St. Louis, M.O.; USA. mPEG2000-PE was synthesized and characterized in house. ALT kit was purchased from Span diagnostic Ltd., Gujarat, India. All other chemicals and solvents were of analytical reagent grade and were used without further purification.
Table 1: Composition, % EE and particle size of freshly prepared liposomal preparation
Types of formulation Lipid Composition % EE (Mean ±SD)
Particle size nm (Mean ±SD)
Drug: Lipid molar ratio
ePC: Chol molar ratio Conventional liposomes (CL) 1:15 7:3 99.28±1.94 136 ±5.8 Sterically Stabilized Liposomes coated with 5 mole % mPEG2000-PE of total lipid (SL) 1:15 7:3 98.54 ±2.34 158 ±6.3
Determination of % Entrapment Efficiency: From each prepared batch, a definite amount of liposomal dispersion was taken and subjected to centrifugation on laboratory centrifuge (Sigma, 3K30) at 15, 000 RPM for 15 min at 0OC after mixing with 50 ml protamine solution (10 mg/ml) using micropipette. The clear supernatant and sediment were separated. The definite amount of supernatant was diluted to 5 ml with methanol and the absorbance was recorded at 295 nm on Shimadzu 1601 UV-Visible Spectrophotometer (Kyoko, Japan) (Schulz et al., 1988). The sediment was resuspended in 1.5 ml distilled water and aliquot was diluted to 10 ml with methanol to lyse the liposomes and the absorbance was recorded at 295 nm. Each time blank containing blank liposome was treated in the same manner to account for any absorbance due to lipid components. Each experiment was repeated three times and average values were calculated (Umrethia et al., 2004).
The entrapment efficiency (% EE) was calculated as follows:
% EE = Amount of FLT in Sediment *100 Equation (1)
Total Amount of FLT added in sample
RESULTS AND DISCUSSION
Characterization of liposomes: Flutamide could be entrapped into CL and SL by thin film hydration technique with the % EE of 99.32% and 98.56%, respectively. The relatively narrow particle size distributions were achieved with an average particle size of CL and SL formulation was 136±5.8 nm and 158±6.3 nm as mentioned in Table 1. The concentration of polymer necessary to produce steric stabilization was determined in vitro by electrolyte-induced flocculation test (Subramanian et al., 2004) at different mole % of mPEG2000-PE and was found to be 5 mole% which gave better stabilization (data not shown).
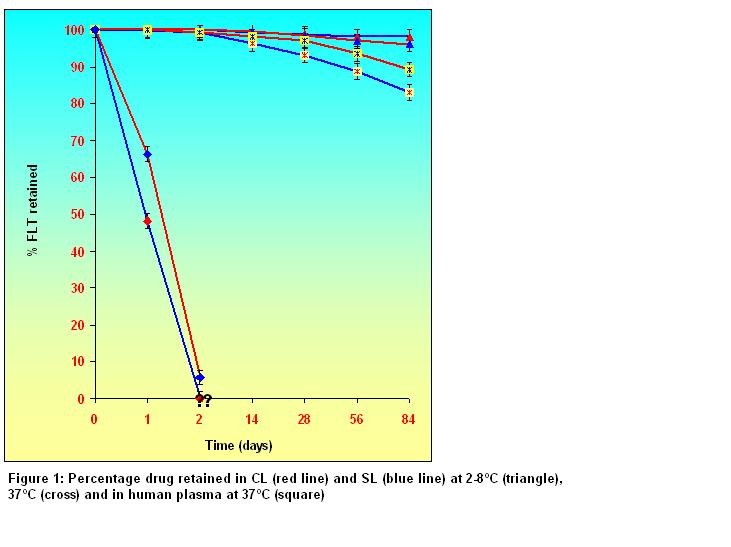
Physical stability of liposomes: Figure 1 shows the % drug retained in lyophilized CL and SL in different conditions. It showed that liposomal preparations were physically stable for more than 3 months in lyophilized form at 2-8°C and retained 90% of their initial content over that period. Leakage of FLT from CL was faster than from SL in human blood plasma at 37ºC. The % drug retained in CL (48.12 %) was less than that of SL (66.38%) after 24 hr and no more drugs was retained after 2 days. Maximum instability was observed at 37ºC in presence of plasma of CL and SL. Thus, from these studies, it was observed that liposomes remained more stable at refrigeration temperature.
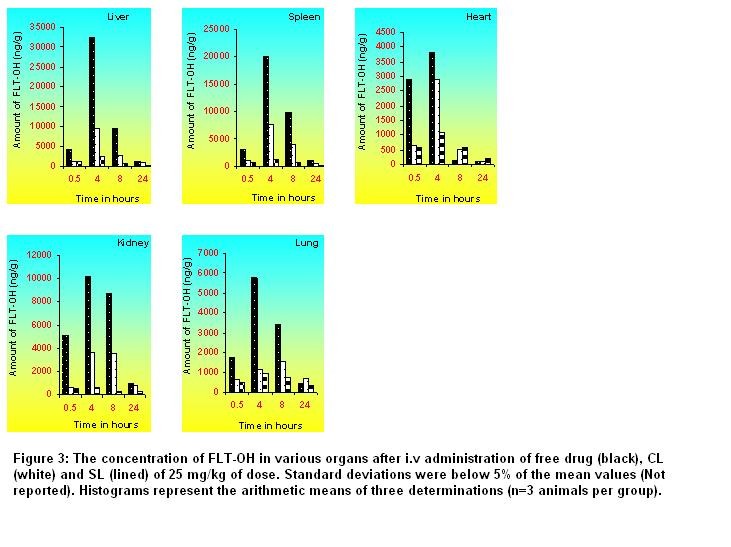
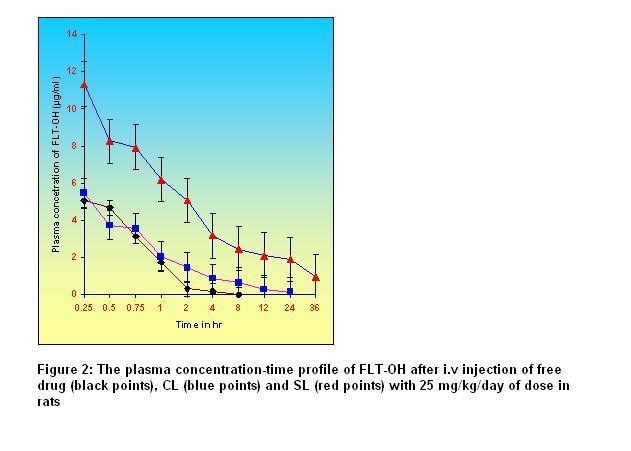
Table 2: One compartmental pharmacokinetic parameters of FLT-OH after i.v administration of free drug, CL and SL containing 25 mg/kg/day of dose to rats*
Pharmacokinetic parameters
SL
CL
Free drug
Cmax (µg/ml)
15.54
7.88
5.48
AUC (µg h ml-1)
85.12
16.41
6.08
AUC∞ (µg h ml-1)
108.72
16.85
6.08
AUMC∞ (µg h2 ml-1)
2536.19
122.42
6.20
MRT (hr)
23.33
7.27
1.02
Clt (l h-1 kg-1)
0.05
0.30
0.82
Vss (l kg-1)
1.15
2.78
1.51
t1/2 (hr)
17.40
6.50
1.28
Kel (/hr)
0.04
0.11
0.54
* The values are arithmetic means of three experiments (n=3 animals per group).
Standard Deviations are less than 5% (Not reported).
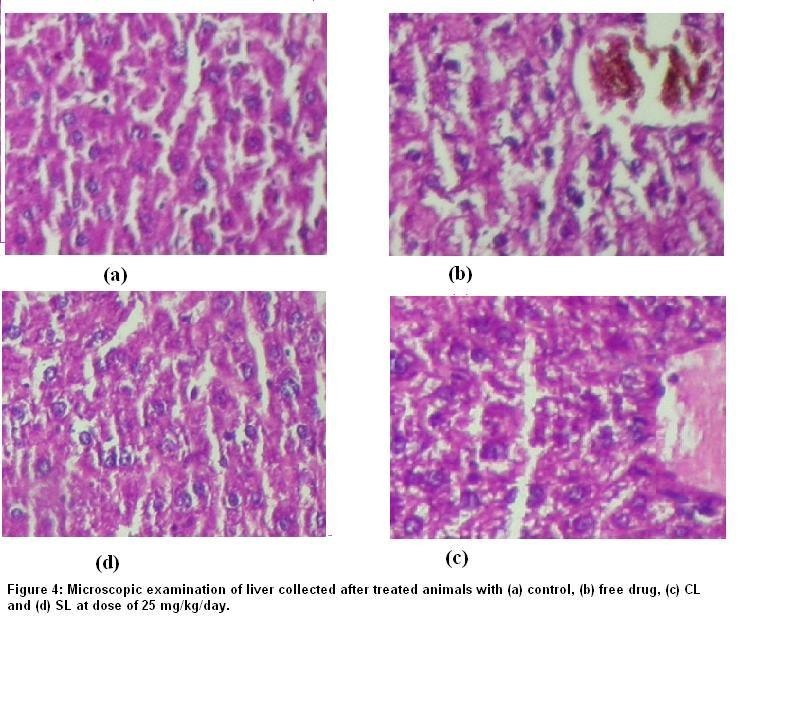
CONCLUSION
The present study showed that conventional and sterically stabilized liposomes with particle size less than 200 nm and high entrapment efficiency can be prepared by thin film hydration technique followed by high-pressure homogenization. The liposomal formulations are stable in lyophilized form and retained enough amount of drug for 24 hr in human plasma. The pharmacokinetic parameters and biodistribution in different organs of sterically stabilized liposomes are significantly different than that of conventional liposomes and the free drug. Sterically Stabilized liposomes exhibited long circulation and fewer uptakes by RES and less change in ALT level, which may help to reduce side effects associated particularly with liver and improve efficacy of the flutamide.
The authors are thankful to TIFAC- CORE for providing the infrastructure, to B.V.Patel PERD Center for HPLC analysis, to Dr. Shailesh Patel (MD) for helping in histopathology and biochemical analysis and to AICTE for financial support as National Doctoral Fellowship to Manish Umrethia
REFERENCES
1. Sharma A and Sharma US. (1997) Liposomes in drug delivery: progress and limitations. International Journal of Pharmaceutics 154:123-140.
2. Labana S and Pandey R. (2002) Chemotherapeutic activity against murine tuberculosis of once weekly administered drugs (isoniazid and rifampicin) encapsulated in liposomes. International Journal of Antimicrobial Agents 20: 301-304.
3. Steerenberg PA and Crommelin DJ. (1984) Reduced cardiotoxicity and nephrotoxicity with preservation of antitumor-activity of doxorubicin entrapped in stable liposomes in the LOU/M Wsl rats. Cancer Research 44: 3698-705.
4. Herman EH and Schein PS. (1983) Prevention of chronic doxorubicin cardiotoxicity in beagles by liposomal encapsulation. Cancer Research 43: 5427-32.
5. Gabizon A and Catane R. (1994) Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene glycol coated liposomes. Cancer Research 54: 987-92.
6. Newman MS and Colbern GT. (1999) Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long circulating, pegylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemotherapy and Pharmacology 43: 1-7.
7. Gondal JA and Preuss HG. (1993) Comparative pharmacological, toxicological and antitumoral evaluation of free and liposome-encapsulated cisplatin in rodents. European Journal for Cancer 29A: 1536-42.
8. Pezer-soler R. (1989) Liposomes as carriers of anti-tumor agents; towards a clinical reality. Cancer Treatment Review 16:67-82.
9. Gabizon A. (1989) Liposomes as a drug delivery system in cancer chemotherapy. In: Drug Carrier Systems, Horizons in Biochemistry and Biophysics Roerdinck F, Kroon A. John Wiley and sons. Vol IX, 185-211.
10. Kondari KS and Suzara V. (2003) Efficacy of liposomal budesonide in experimental ashthma. Journal of Allergy and Clinical Immunology 111 (2):321-327.
11. Dijkstra J and Kalicharan D. (1984) Interaction of liposomes with Kupffer cells in vitro: Experimental Cell Research. 150:160-176.
12. Woodle MC. (1998) Controlling liposomes blood clearance by surface grafted polymers. Advanced Drug Delivery Review 32:139-152.
13. Goren D and Lossos A. (1997) Long-circulating liposomes for drug delivery in cancer therapy: A review of biodistribution studies in tumor-bearing animals. Advanced Drug Delivery Review 24:337-344.
14. Lasic DD. (1995) The stealth liposomes: a prototypical biomaterial. Chemical Review 95:2601-2634.
15. Torchilin VP. (1995) Long-circulating drug delivery systems. Advanced Drug Delivery Review 16:125-348.
16. Allen T and Mathhay K. (1991) Sterically stabilized liposomes improvements in pharmacokinetics and anti-tumor therapeutic efficacy. Proceedings of the National Academy of Sciences USA. 88:11460-11464.
17. Oku N and Namba Y (1994) Long-circulating liposomes. Critical Reviews in Therapeutic Drug Carrier Systems 11: 231-274.
18. Baker JW and Bachman, GL. (1967) Synthesis and Bacteriostatic Activity of Nirtrofluromethylenilides. Journal of Medicinal Chemistry. 10:93-95
.
19. Goldspiel BR and Kholer DR. (1990) Flutamide: an antiandrogen for advanced prostate cancer Drug Intelligence and Clinical Pharmacy 24:611-623.
20. Chabner BA and Calabresi P. (1996) in: The Pharmacological Basis of Therapeutics. A. Goodman, J.G. Hardman, L.E. Limbird, P.B. Milinoff, R.W. Ruddan , 9th ed., Mc-Graw Hill, Mexico, 1309-1367.
21. Umrethia ML and Murthy RSR. (2004) Optimization of formulation parameters of flutamide liposomes by 33 factorial 26-term logit model. Pharmaceutical Development Technology 9(4):369-378.
22. Schulz M and Schmoldt A. (1988) The Pharmacokinetics of Flutamide and Its Major Metabolites after a Single Oral Dose and During Chronic Treatment. European Journal of Clinical Pharmacology 34:633-636.
23. Dosio F, Brusa P, Crosasso P, Arpicco S. (1997) Preparation, Characterization properties, in vitro and in vivo of paclitaxel albumin conjugate. J. Control. Release 47:293-304.
25. Subramanian N and Murthy R.S.R. (2004) Use of electrolyte induced flocculation technique for in-vitro steric stability of sterically stabilized liposomal formulation. Die Pharmazie 1:69-74.
26. Allen TM. (1998) Liposomal drug formulations- rationale for development and what we can expect from the future? Drugs 56(5):747-756.
27. Gabizon A and Catane R. (1994) Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene glycol coated liposomes Cancer Research 54 (4):987-992.
28. Liu DX and Liu F. (1995) Recognition and clearance of liposomes containing phosphotidylserine are mediated by serum opsonins. Biochimica et Biophysica Acta-Biomembranes 1235:140-146.
29. Efremova NK and Bondurant B. (2000) Measurements of interbilayer forces and protein adsorption on uncharged lipid bilayers displaying poly (ethylene glycol) chains. Biochemistry 39:3441-3451.
30. Woodle MC. (1993) Surface-modified liposome-assessment and characterization for increased stability and prolong blood circulation. Chemistry and Physics of Lipids 64(1-3):249-262.
31. Lee JCM and Bermudez H. (2001) Preparation, stability and in-vivo performance of vesicles made with diblock copolymers. Biotechnology and Bioenergetics 73 (2):135-145.
32. Gabizon G and Papahadjopoulos D. (1988) Liposomes formulation with prolonged circulation time in blood and enhance uptake by tumors. Proceedings of the National Academy of Sciences USA. 85:6949-6953.
33. Ozonol S and Kawakami1 T. (2002) Caffeine test in predicting flutamide-induced hepatic injury in patients with prostate cancer. Prostate Cancer and Prostatic Diseases 5:128–131.
34. Huang L. (1992) Covalently attached polymers and glycans to alter the biodistribution of liposomes. Journal of Liposome Research 2:289-291.
35. Kim CK and Han JH. (1994) Pharmacokinetics and tissue distribution of methotrexate after intravenous injection of differently charged liposome-entrapped methotrexate to rats. International Journal of Pharmaceutics 108:21-29.
36. Oku N. (1999) Anti-cancer therapy using glucuronate modified long-circulating liposomes. Advanced Drug Delivery Review 40:63-73.
©1996-2005 All Rights Reserved. Online Journal of Pharmacokinetics. You may not store these pages in any form except for your own personal use. All other usage or distribution is illegal under international copyright treaties. Permission to use any of these pages in any other way besides the before mentioned must be gained in writing from the publisher. This article is exclusively copyrighted in its entirety to OJPK publications. This article may be copied once but may not be, reproduced or re-transmitted without the express permission of the editors.